





Abstract
Objective: Although trait-associated genes identified as complex versus single-gene inheritance differ substantially in odds ratio, the authors nonetheless posit that their mechanistic concordance can reveal fundamental properties of the genetic architecture, allowing the automated interpretation of unique polymorphisms within a personal genome.
Materials and methods: An analytical method, SPADE-gen, spanning three biological scales was developed to demonstrate the mechanistic concordance between Mendelian and complex inheritance of Alzheimer's disease (AD) genes: biological functions (BP), protein interaction modeling, and protein domain implicated in the disease-associated polymorphism.
Results: Among Gene Ontology (GO) biological processes (BP) enriched at a false detection rate <5% in 15 AD genes of Mendelian inheritance (Online Mendelian Inheritance in Man) and independently in those of complex inheritance (25 host genes of intragenic AD single-nucleotide polymorphisms confirmed in genome-wide association studies), 16 overlapped (empirical p=0.007) and 45 were similar (empirical p<0.009; information theory). SPAN network modeling extended the canonical pathway of AD (KEGG) with 26 new protein interactions (empirical p<0.0001).
Discussion: The study prioritized new AD-associated biological mechanisms and focused the analysis on previously unreported interactions associated with the biological processes of polymorphisms that affect specific protein domains within characterized AD genes and their direct interactors using (1) concordant GO-BP and (2) domain interactions within STRING protein–protein interactions corresponding to the genomic location of the AD polymorphism (eg, EPHA1, APOE, and CD2AP).
Conclusion: These results are in line with unique-event polymorphism theory, indicating how disease-associated polymorphisms of Mendelian or complex inheritance relate genetically to those observed as ‘unique personal variants’. They also provide insight for identifying novel targets, for repositioning drugs, and for personal therapeutics.
Background and significance
Alzheimer's disease (AD) is the most common type of dementia, characterized by a severe form of memory loss and deterioration of other cognitive functions. It currently affects 30 million people worldwide, and this number is expected to quadruple by 2050.1 Great strides have been made in AD research to unveil genetic underpinnings and provide a foundation for a personal genomics solution to treat the disease. Preceding the findings of common variants in ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP by Hollingworth et al in their recent genome-wide association study (GWAS, GERAD+) involving over 50 000 patients, other GWAS have identified a total of 19 genes encompassing single-nucleotide polymorphisms (SNPs) associated with an increased risk of developing AD.2 Additionally, 15 Online Mendelian Inheritance in Man (OMIM) genes showing single-gene inheritance associated with AD have been annotated (http://www.ncbi.nlm.nih.gov/omim). Polymorphisms in the APOE gene are the most well documented genetic risk factors for developing early-onset AD.3,4 In particular, APOE4 accounts for 50% of cases and has been found to increase the relative risk for early-onset AD in line with its allelic prevalence—E4/E4 (14.9), E3/E4 (3.2), E2/E4 (2.6), and E2/E2 (0.6); conversely, APOE2 appears to have a protective effect against AD.5,6 With the accumulation of new genetic insights into AD, systems biology and systems medicine approaches are poised to derive new meanings from interactions among genetic variants.
Following these recent developments in AD GWAS, we identified a large void in the interpretation and integrative capability of higher scales of biology within these susceptibility loci. Namely, previous reductionist genetic approaches have not been able to sufficiently indicate AD as a complex disease. Indeed, the pathogenesis of sporadic AD has been widely attributed to both genetic and environmental factors, while pure autosomal dominant Mendelian transmittance accounts for a smaller proportion of cases (10%).7,8 While APOE, APP, PSEN1 and PSEN2 have been characterized as true deterministic genes, other genetic loci increase the risk of developing AD. Thus, being able to characterize the connectivity and directionality of the relationships between the underlying genetics and corresponding functions in high-throughput data may break boundaries between specialized silos of knowledge of gene functions and greatly enhance a holistic approach to understanding the disease. Furthermore, substantive efforts have yet to be made to investigate the functional overlap—of relevance to a personal genome—between AD genes showing classical Mendelian and complex modes of inheritance.
The presentation of the seminal evaluation of incorporating personal genome information into a modern clinical assessment by Ashley et al demonstrated the powerful utility of integrating common polymorphisms to determine risk of disease within a single patient.9 However, the clinical relevance of the majority of ‘unique personal genetic variants’ remains unrecognized.9 Furthermore, aggregating the significance of these unique personal variants within a patient along with polymorphisms known through disparate modes of inheritance and non-genetic factors may provide key knowledge about an individual's risk and pathology of disease. For instance, despite the increased risk and higher OR of AD attributed to APOE4, many E4/E4 homozygotes live to old age with no indication for AD, and up to 50–75% of heterozygotes carrying one E3 allele never develop AD.6 However, numerous studies have established that Aβ deposition in the brain and poorer outcome in terms of neurodegenerative disease after head injury occur more commonly in individuals possessing an APOE4 allele.6,10 Therefore, the APOE4 variant represents a paradigmatic example of complex inheritance of AD in its intersection between genetic predisposition and a plausible environmental factor associated with the disease.
In traditional GWAS, biological function is inferred from a small set of sequence elements within loci, and they require multiple patients to establish a prediction. To date, no predictive methods have been applied to establish the association between a trait and unique personal variants in an uncharacterized gene or in uncharacterized polymorphisms of a gene harboring disease-associated polymorphisms. In the past, reverse genetic methods have predicted gene function from molecular similarity of sequence or structure, and could in theory be applied to prediction of unique personal genomic variants. Conversely, forward genomics examine higher systems properties of high-throughput data and subsequently zoom in on causal genetic roots. Such forward genomics techniques have also proven successful for arriving at new phenotype-associated variants in a variety of contexts.11,12 For instance, we have shown that the same systems properties of biological processes and molecular functions are consistently enriched among the top 1000 genes of independent adult-onset diabetes mellitus.13 Previously, our group has also shown that properties at the protein-interaction level are able to establish overlap between diseases and predict novel candidates involved in molecular mechanisms of disease.14,15 Additionally, Zhong et al demonstrated that specific edgetic alleles (mutations responsible for specific protein-interaction patterns) are associated with certain Mendelian diseases, as compared with other edgetic or null alleles (mutations responsible for structural alteration and complete loss of protein interactions).16 Further, it has been shown by our group and others that disease trait similarity of complex diseases can be imputed from genetic variants.17,18 Here we demonstrate the relevance of edgetic properties of Mendelian and complex disease inheritance genes in AD, as well as integrate a forward genomics approach to arrive at new hypotheses for risk inheritance. Analyzing SNPs at the mechanism level of the gene also addresses the current limitation of GWAS described by Goldstein's group—SNPs may be markers of rare or unique personal variants, as opposed to the prevailing belief that they measure a nearby common variant with minor allele frequency >5% (consensus definition of a SNP).19
We thus hypothesized that the clinical significance of unique personal variants could be imputed with increased accuracy by triangulating three established approaches: forward genomics, reverse genetics, and computational biology modeling of systems and networks (online supplementary figure S1). Lee et al and other groups have established the proof of concept for using Gene Ontology (GO) similarity and protein interactions to prioritize genes associated with a disease.20–22 Yet, none of these studies have investigated similarity between disease traits or similarity between polymorphisms according to their mode of inheritance (single gene vs complex). Here we analyze 40 genes shown to be associated with AD, using text-mining techniques to characterize mechanistic commonalities and inter-relationships between GO biological processes (GO-BP) enriched within SNPs from OMIM and confirmed in GWAS (online supplementary tables S1 and S2). We have previously demonstrated the feasibility and utility of a novel information theory-based method for predicting protein functions and building disease–disease networks by exploiting the semantic similarity of GO terms among host genes of validated trait-associated SNPs.13 We apply this forward genomic method of GO term enrichment and scoring of AD SNPs based on information theory semantic similarity (ITSS) scores which we developed23 in order to construct the functional space of AD polymorphism host genes. We further constrain our predictive space by examining the node and edgetic significances within protein-interaction networks (PINs) and domain–domain interactions of AD genes and canonical pathways. Taken together, our mechanism-guided approach to integrating intermediate phenotypes derived from forward genomics lays a foundation for translating unique personal variants into other established networks used for drug repositioning (figure 1).
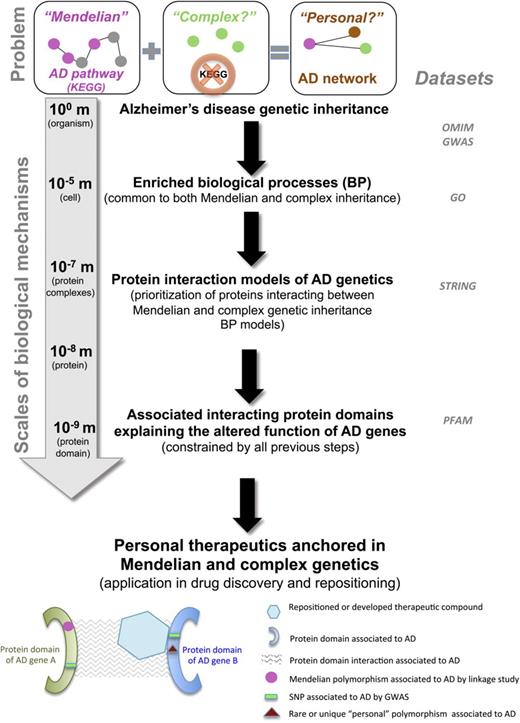
Scalar protein analysis of domains enriched in genetics (SPADE-gen): changing the paradigm of drug repositioning for complex diseases with genetically anchored biological mechanisms. The diagram shows the stepwise process by which the proposed SPADE-gen method identifies concordance between Mendelian and complex disease genetics and represents it as nested mechanisms which recapitulate their genetic biology at multiple scales: (1) biological processes associated with Alzheimer's disease (AD); (2) protein interactions involved in these biological processes; (3) their associated interacting protein domains which may explain the altered function of inheritable AD genes. Finally, this multiscale knowledge is applied to implicate new, rare, or ‘unique and personal’ polymorphisms within these AD-associated protein domains. In summary, this method translates genetic signals into the protein domain language required for use in biological and computational drug repositioning pipelines which cannot, by design, directly incorporate complex disease genetics. Datasets used by this method are listed in the right column of the figure. The problem statement is outlined at the top of the figure and is as follows: while Mendelian AD genes are well understood in established AD molecular pathways (eg, KEGG), the host genes of over 20 newly discovered intragenic AD single-nucleotide polymorphisms are surprisingly neither part of the AD pathways nor direct interactors with these proteins. The base of the figure illustrates the intended utilization: a therapeutic compound repositioned or developed specifically to interact within an AD-associated protein domain.
Materials and methods
Data
A set of eight somatic SNPs discovered in two recent AD GWAS meta-analyses, ADGC24 and GERAD+,2 were included in our study. Another 17 AD-associated SNPs were identified from the GWAS catalog of association loci for human diseases and traits downloaded from the National Human Genome Research Institute (NHGRI) website (http://www.genome.gov/gwastudies/ Apr 2011) (NHGRI GWAS Catalog). KEGG AD and Parkinson's disease pathways were obtained from the KEGG website (http://www.genome.jp/kegg/pathway/hsa/hsa05010.html). The KEGG database was downloaded through R package ‘KEGG’. The GO terms enrichment package, GOstats, was download from the Bioconductor website (http://www.bioconductor.org/packages/2.8/bioc/html/GOstats.html). To identify true findings from GO enrichment, we used a web-accessible tool, GO-module (http://www.lussierlab.org/GO-Module). OMIM genes' background used in permutations was downloaded from the Human Genome Organizations (HUGO)'s Gene Nomenclature Committee (http://www.genenames.org/cgi-bin/hgnc_downloads.cgi June 23, 2011). All information on single inheritance genes associated with AD was obtained from the OMIM website (http://www.ncbi.nlm.nih.gov/omim). The gene to GO terms relational database was downloaded from the NCBI website (http://www.ncbi.nlm.nih.gov/entrez/query/static/help/LL2G.html#files) via FTP. The protein interactions were downloaded from the search tool for the retrieval of interacting genes (STRING) version 8.0 on December 19, 2008 (http://string.embl.de).25
AD GWAS SNP's host genes and OMIM genes
In this study, we used two recently published GWAS: The Alzheimer's Disease Genetics Consortium26 and GERAD+ consortia (including data from the GERAD,27 EADI,28 Translational Genomics Research Institute (TGEN1)29 and the Alzheimer's Disease Neuroimaging Initiative)30 and all SNPs found in previous AD GWAS. We included a total of 25 AD genes of complex inheritance annotated to the reported SNPs in both papers (closest to the SNPs) and the NHGRI GWAS catalog. Sixteen Mendelian inheritance AD genes were identified in the OMIM using text mining, which we previously published.31 One overlapping gene (APOE) was found in both GWAS and OMIM lists. In other words, distinct polymorphisms of APOE are responsible for the high OR and penetrance of the Mendelian inheritance alleles reported in OMIM and for the low OR and penetrance of the GWAS SNP associated with complex inheritance. To best illustrate the power of our system's concordance analysis, we applied a rather conservative method and removed APOE from the OMIM gene list in order to generate distinct and independent gene lists for GWAS and OMIM.
Functional annotation of GWAS and OMIM genes to GO
To find significant genes in both of our gene lists, we first conducted KEGG canonical pathway enrichments between GWAS genes and between OMIM genes using four KEGG pathways associated with AD: AD (hsa05010), apoptosis (hsa04210), calcium-signaling pathway (hsa04020), and oxidative phosphorylation (hsa00190). We also conducted a GO enrichment study to prioritize biomolecular systems related to AD using genes annotated to GWAS SNPs and those identified in the OMIM dataset. The unadjusted p value of the GO enrichment was calculated using the cumulative hypergeometric distribution provided by an open source R package (GOstats, Bioconductor),32 with the parameter ‘conditional on the GO structure’. Benjamini–Hochberg correction33 was applied to control for multiple comparisons. To prioritize and refine the enrichment, we filtered the enriched GO terms using a web-accessible tool that we developed, GO-module,34 which reduces the GO complexity by constructing biomodules from significant GO terms based on hierarchical knowledge.
ITSS between GO terms enriched from the gene list of complex inheritance (GWAS) and those from Mendelian disorders (OMIM)
To pinpoint the common systems emerging from complex (GWAS) and single-gene (OMIM) inheritance of AD, we used a previously implemented algorithm to calculate ITSS (range 0–1) between GO terms and only included ITSS score ≥0.7 (which we have previously shown to be significant in optimizing systems prediction).23 GO terms enriched in each GWAS were systematically compared with one another using (1) simple overlap (eg, same GO terms or ITSS=1), and (2) with GO terms with ITSS ≥0.7 (see equations 1 and 2). One thousand bootstraps were conducted for the 15 OMIM genes and subjected to the same analyses (GO enrichment, GO-module refinements, and ITSS between GO terms from the OMIM bootstrap and those from the GWAS enrichment). GWAS and OMIM enrichment recalls were conducted by the same GO enrichment methods. Subsequent ‘leave one gene out’ analyses using the same methods were conducted to verify the robustness of the GO terms enriched in either the GWAS or OMIM gene list.
Genome sequence of individuals with AD
Queries were made across all PubMed databases, http://alzforum.org/, and Google-powered searches.
Protein family motif (Pfam) annotations
Non-synonymous SNPs in our case study network containing two overlapping GO terms (false discovery rate (FDR) <5%) were manually analyzed for associated protein family domains (Pfam). We queried the Ensembl Genome Browser (http://www.ensembl.org/) for transcript and gene level information for all missense SNPs in our dataset, and then confirmed existing Pfam domains encompassing each SNP from Ensembl protein IDs. Intronic SNPs were analyzed by searching for an associated exon coding sequence encompassing each intronic SNP within the UCSC Genome Browser (http://genome.ucsc.edu/) to obtain a protein sequence, and subsequently determined Pfam domains from Ensembl protein IDs.
Linking protein domains to OMIM, GWAS, and Parkinson's disease genes
We mapped all Pfam36 domains linked to the corresponding gene in GWAS, OMIM, and Parkinson's disease datasets. In order to map domains to proteins, HMMer's semi-global implementation37 was used to search for complete domains in human proteins as shown in previous work for the Domain Mapping of Disease Mutations database.38
Construction of domain–domain interaction networks
In order to create domain–domain interaction networks, we retrieved theoretical predictions for GWAS, OMIM, and Parkinson's disease genes from the DOMINE database (version 2.0 released September 2010).39 GWAS and OMIM genes were then analyzed for shared domain–domain interactions in their first- and second-degree domain–domain interaction networks. To find the first- and second-degree interaction networks for the GWAS and OMIM genes, we used protein–protein interactions from the STRING database with a combined score greater than or equal to 900 for each of the GWAS and OMIM genes. Predictions of domain–domain interactions were based on experimental data on protein interaction and used the relative frequency of interacting domains,40 maximum likelihood estimation of domain interaction probability,41,42 or network properties to predict protein–domain interactions.43,44 We compiled domain–domain interaction predictions from all these methods from the DOMINE database. Using these predictions, we found theoretical domain–domain interactions between all pairs of proteins in first- and second-degree networks for the GWAS and OMIM genes. The first-degree theoretical domain–domain interactions for GWAS genes were compared against the first-degree interactions for OMIM genes to find overlapping Pfam domains. We created a network using domain–domain interaction data for GWAS and OMIM AD genes and the maximum biological process ITSS score using Cytoscape.45
Construction of protein–protein interaction networks and network topology for AD and Parkinson's disease
We used a similar method to extract interactions from STRING as previously introduced by Chen et al.46 Distinct interactions between 40 AD proteins and all proteins from four AD KEGG pathways were retained. Negative control tests were conducted between the proteins from Crohn's disease, epithelial cancers, or breast cancers and those from the four AD KEGG pathways. Interaction significances (p values) were calculated at edgetic level using the Fisher exact test. We calculated the total potential edges from the available proteins in the PIN. Expected edges were calculated between proteins from AD or Parkinson's disease and those from each pathway we used. In the networks, nodes represent proteins, and edges represent interactions between proteins. Node hubness and bottleneckness, as well as edgetic significances, were used to prioritize the protein interactions (SPAN31). Hubness was calculated as the highest 20% ranked genes according to the node degree calculated from the filtered STRING PIN. Bottleneckness was calculated using Gerstein's laboratory software, and a rank <20% corresponds to high betweenness.47 Comparisons of genes' hub and bottleneck ranks were conducted using the non-parametric Mann–Whitney test (two-group comparison) and Wilcoxon signed rank test (comparison with theoretical median of 50%).
Results
Intermediate phenotypes predicted by exact overlaps and ITSS of GO enrichment
We obtained 25 complex trait inheritance SNPs from previous GWAS (25 genes) and 183 allelic variants from single-gene inheritance (15 genes) from OMIM. We note that, although APOE is annotated in both AD GWAS and OMIM genesets, we prioritize it as an OMIM gene in this study for its greater OR as a Mendelian gene in this study. As a preliminary study, we incorporated the GWAS variants with the OMIM variants and compared them with known AD pathways (KEGG). Using canonical pathways from the manually curated KEGG database, we found no more than five overlaps between the OMIM genes and any of the four pathway genesets associated with AD. Moreover, the overlap of this straightforward approach is not statistically significant after FDR adjustment (data not shown). Further, there is no overlap between any of the canonical pathway genesets and the GWAS genes, indicating the lack of sensitivity of this simple pathway-based enrichment analysis. To identify the systems medicine properties of single-gene and complex inheritance in AD that could be of use in personal genomics, we first approached these questions from a higher, systems (GO), level. However, even at a higher GO level, the genetic structures of AD can be extremely complex: systems biology properties of AD are buried among 729 GO terms associated with the 40 identified AD genes and 2281 other genes annotated to these GO terms.
By calculating the empirical probabilities of (1) the exact overlaps (figure 2A, online supplementary figure S2A) and (2) similarities (figure 2B, online supplementary figure S2B) between GO-BPs enriched in the complex inheritance (GWAS) and those enriched in single-gene inheritance (OMIM), we are able to prioritize known molecular mechanisms of AD and potentially identify new ones. Figure 2A provides the number (two matches of 22 enriched GWAS GO terms and 37 enriched OMIM GO terms) and observed p value (0.003) of overlapping GO-BPs enriched between GWAS and OMIM genes after adjustment for the GO enrichment p values for multiple testing and with a cut-off at an FDR of 5%. In addition to exact overlapping GO terms, we also used GO annotations to identify similar intermediate phenotypes, which were then calculated by ITSS metrics (Materials and methods). Between 22 GO terms enriched in GWAS and 37 enriched in OMIM (online supplementary tables S3 and S4), we identified 45 ‘similar pairs of GO-BPs’ among 814 potential pairs using a stringent criterion (ITSS >0.7; Materials and methods) that we have previously shown to be optimal for the precision and recall of this similarity approach.23 The observed p value for the average ITSS score among 814 pairs (0.119) is 0.009. Figure 2C provides the network of similarity-predicted GO terms enriched between the two studies according to the same parameters as figure 2A,B. We also calculated the observed p value for exact matches and ITSS score using unadjusted p values <0.05. We found 16 exact matches with observed p value 0.007 and an average ITSS score of 0.32 with observed p value <0.001 (online supplementary figure S2A,B). These results confirmed our finding at a more relaxed cut-off level, indicating the scalability of our approach. We also confirmed the robustness of these results by systematically leaving each AD gene out of the enrichment one at a time (Materials and methods; data not shown).
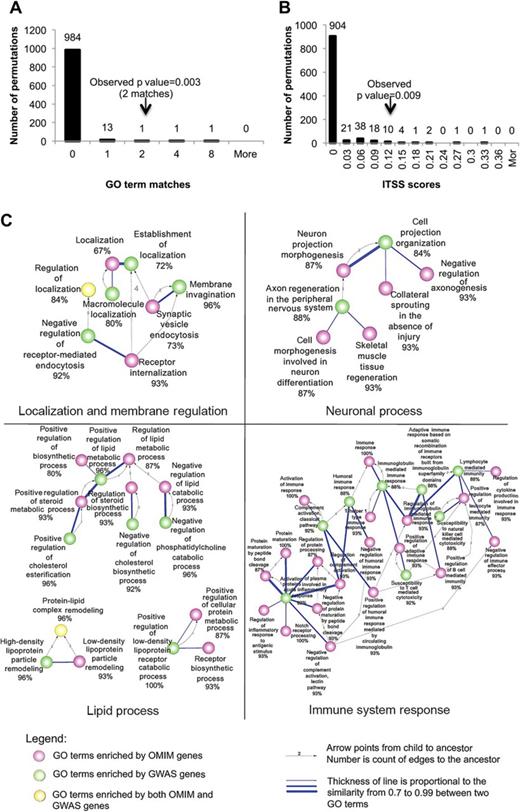
Overlap and similarity networks between Gene Ontology (GO) biological processes enriched in single-gene (Online Mendelian Inheritance in Man (OMIM)) and complex (genome-wide association studies (GWAS)) inheritance Alzheimer's disease (AD) genes. Empirical distributions were conducted by bootstrap (Materials and methods) to derive the p value of the observed exact overlap of GO terms enriched between the OMIM genes and those of the GWAS at a false detection rate (FDR) <5% cut-off of enrichments (A). Using information theoretic semantic similarity (ITSS, Materials and methods), a similar empirical calculation was conducted to identify similar GO terms enriched between the studies at the same cut-off of enrichments (B). Each bar presents the empirical distributions (arrows point to the observed results). This study confirms that specific biological processes underpin the pathophysiology of AD regardless of the mode of inheritance. GO terms were found to be enriched for 15 OMIM and 25 GWAS genes using stringent similarity and FDR criteria of (A) and (B), and, of 45 pairs found to be similar, (C), four biomodules were identified: localization and membrane regulation (eight GO terms); neuronal process (10 GO terms); lipid process (12 GO terms); immune system response (27 GO terms).
Network of similar GO-BPs enriched in both Mendelian and complex AD inheritance
It has been shown that ITSS scores can predict biomolecular systems properties.13,23 A total of 45 pairs of GO terms were identified as significantly similar pairs (ITSS >0.7) from GWAS and OMIM enrichment with an FDR <5%. From these 45 pairs, a network of intermediate phenotypes with significant substructures emerged. By connecting similar pairs with weighted lines indicating the level of similarity, clusters of significant GO terms were identified representing intermediate phenotypes (figure 2C). Among the connections between similar pairs of GO terms, we identified four significant biomodules of GO-BPs describing established functional aspects of AD according to our review of the literature (online supplementary table S5). In principle, each of these biomodules could organize unique combinations of single-gene and complex-gene inheritance of AD within an individual.
PIN of known AD KEGG pathway genes with GWAS and OMIM AD genes
Owing to the significant similarity observed at the intermediate (GO-BP) phenotype level of the OMIM and GWAS genes, we were able to combine the two sets of genes and prioritize them according to known PINs (STRING v8.0), although the small number of overlapping GO terms limited our power to predict future variants. In order to impute the significance and functions of future variants, we first constructed the PIN between the proteins of the 40 AD genes and the proteins from the known KEGG AD pathway (hsa05010) (figure 3). Based on our empirical distributions, we prioritized edges in the PIN based on both the nodes' connectivity and their edgetic significances using the SPAN network model that we developed31 (Materials and methods). As shown in figure 3, we identified one significant connection between a GWAS and KEGG gene, CLU and APP, respectively. We also found three significant proteins from GWAS (PICALM, CLU, and BIN1), seven proteins from OMIM (A2M, APP, PSEN1, PSEN2, PRNP, SNCA, and NOS33), and the overlapping APOE gene after adjustment of p values with controlled empirical simulations. Twelve proteins identified in GWAS were not found to be in the network because of a lack of protein function data. While no AD gene of complex inheritance discovered by GWAS was represented in KEGG, this study shows that conservative network models can identify these genes as statistically significant first interactors with known AD proteins from KEGG; in other words, molecular mechanisms can be imputed for newly discovered GWAS genes with known biology of the disease.
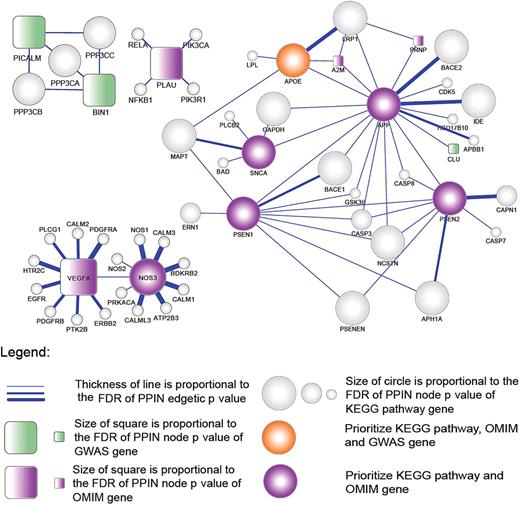
Protein-interaction network (PIN) between 40 Alzheimer's disease (AD) genes of complex and Mendelian inheritance and KEGG AD pathway genes. Of the 40 AD inheritance genes, 28 are connected through protein–protein interactions in the network using a threshold cut-off of 900 within the STRING database and stringent network modeling using SPAN (Materials and methods). Node and edgetic significances, representing individual proteins and their interactions, respectively, are visualized according to their respective false detection rate (FDR) p value. Node shape and color indicate the source of genes: circle (contained by KEGG), square (not contained by KEGG), gray color (KEGG only), green (genome-wide association studies (GWAS)), purple (Online Mendelian Inheritance in Man (OMIM)), and orange (OMIM and GWAS).
Previous studies investigating the biological role of network topology of PINs have shown that network hubs are associated with disease genes, and bottlenecks often correspond to dynamic functional components.48 We observed that bottleneckness and hubness of Mendelian (OMIM) AD genes are significantly increased compared with those of a random selection of genes from the network (figure 3; lower rank of betweenness and node degree correspond to increased bottleneckness and hubness, respectively; p<0.0001 in comparison with the random selection; Wilcoxon signed ranked test in comparison with the median rank of network genes). In addition, AD genes with Mendelian inheritance showed more bottleneckness and hubness than those of complex inheritance discovered by GWAS (figure 3; p=0.0007 and p=0.088 respectively, Mann–Whitney test). According to the network topology, APP, PSEN1, PSEN2, and SNCA were identified as hubs as well as bottlenecks from the interaction network between KEGG AD pathway and candidate genes. As shown in figure 3, for example, two GWAS genes (BIN1 and PICALM) are connected through three KEGG pathway proteins (PPP3CA, PPP3CB, PPP3CC), and BIN1 is among the top nodes in terms of bottleneckness and hubness. Further PIN analysis between our candidate genes, with the KEGG AD (hsa05010) and KEGG apoptosis (hsa04120) pathway, also confirmed the priority of BIN1 and PICALM (data not shown). In addition, PLAU was also prioritized with significant bottleneckness in the interaction network with the KEGG AD pathway. Similarly, through interactions with the KEGG calcium-signaling pathway, VEGFA and NOS3 were identified as significant hubs and bottlenecks. Thus, with network topology analysis, we can further suggest the significance of AD GWAS playing a role in established pathological pathways of AD.
Discussion
Comprehensive approaches to analyzing personal genomes in concert with rare variants of disease remains a major challenge for systems and computational biology.9 Being able to characterize the mechanistic overlap between single and complex gene inheritance may allow improved assessment of an individual's risk of disease, as unique personal or rare variants contributing to the phenotype are likely to occur within those mechanisms as well. In our combined top-down forward genomics and bottom-up network modeling approach, we use four sources of knowledge (KEGG pathways, GO enrichment, protein–protein interaction networks, and protein–domain interactions) with AD genes obtained from OMIM and recent GWAS in order to impute genomic regions in new risk variants within personal genomes.
Geneset enrichment using canonical KEGG AD pathways versus unbiased GO classes
The failure to enrich the complex disease AD genes derived from GWAS or well-established Mendelian inheritance AD genes derived from OMIM in any one of the four known KEGG pathways associated with AD was expected because of: the small number of genes in each set; the inclusion of deregulated AD genes discovered in cell signaling and biological experiments, rather than through genetic evidence, in the KEGG database; and, significantly, the fact that new discoveries from GWAS may comprise uncharacterized biological mechanisms not yet associated with the ‘canonical’ pathways agreed to be associated with AD pathogenesis. These results provided the rationale for a broader unbiased assessment of all genesets annotated in GO. Indeed, it has been shown in complex inheritance signals from GWAS that (1) multiple intragenic SNPs prioritized in GWAS can yield a statistically significant joint mechanism defined as a GO geneset49,50 and (2) a GO overlap signal can be identified among different GWAS that otherwise have no gene overlap.14 Unsurprisingly, we report shared and similar GO terms enriched between host genes of intragenic SNPs reported in GWAS with monogenic AD trait genes, as well as those with high OR annotated in OMIM. However, different from these previous studies which were conducted over hundreds of signals from single GWAS, here the intragenic SNPs and OMIM genes had all been confirmed in repeated studies, and thus we postulate that they represent a focused system of intermediate phenotypes that define commonalities between otherwise heterogeneous molecular presentations of AD. These unbiased GO mechanisms that we report to be associated with both complex and monogenic inheritance of AD are further constrained by another biological scale: PINs.
Protein-interaction modeling
Despite constraining our network down to top-most interactions, we could still find several interesting patterns. Seven genes not included in the KEGG pathway were prioritized using PINs. Among them, three were GWAS genes with significant bottleneckness and hubness. In addition to significant genes identified from the PINs, we also found significant edges that connect AD GWAS or OMIM proteins and KEGG pathway proteins. Therefore, by constructing PINs, we were able to impute the function and significance of several variants by their node and edgetic properties. One constraint on the predictive power of the PIN is that only very high quality protein interactions from the STRING database were included. We can expand our model and impute more variants with larger amounts of information by including interactions from a broader range of quality-control cut-offs. Further, when combining protein–protein interaction information, as well as structural information (ie, splice sites, promoters, or Pfams), with GO terms associated with a new variant, we can even pinpoint the possible function of the new variants. Because of the edgetic significance obtained from PINs, we can also impute significant variants from the first interaction partners of the protein. An additional analysis of Parkinson's disease overlap through protein–protein interactions can be found in online supplementary table S6.
Shared Pfam analysis
SNPs identified from OMIM AD genes were also found to be connected to a number of other diseases, including Parkinson's, hyperlipoproteinemia, lipoprotein glomerulopathy, and myeloperoxidase deficiency (online supplementary table S2). Interestingly, we found that heterogeneous disease associations for SNPs within each gene converged on the same Pfam when we mapped SNPs to known Pfams from the Ensembl database (data not shown). These findings suggest that our forward phenomics approach may be more insightful than a reverse genetics approach for predicting new rare variants, as a reductionist approach initially constrained at the Pfam level may be biased to predicting incorrect disease associations. Further, as Goldstein's group has demonstrated, many SNPs discovered in GWAS may be markers of rare or personal variants rather than the prevailing belief that a local frequent allele is responsible for the statistical signal.19 Notably, intragenic SNPs of a particular gene were only associated with AD in a few cases, which could increase the chance that additional novel variants may associate with incorrect disease traits. Taken together, these results suggest that (1) a forward genomics approach triangulating on mechanisms responsible for the disease using single- and complex-gene inheritance should be conducted initially, (2) only the biologically validated Pfam or SNPs be used to guide PINs, and (3) bioinformatics modeling of the rare variants associated with the GWAS SNPs need to be incorporated in addition to deep sequencing of these regions.
Integrating intermediate phenotype similarity with domain–domain interactions
Progress has been made in recent efforts to integrate intermediate phenotypes with clinical and molecular phenotypes in many disease contexts.51 We show in figure 4 that, indeed, constraining analysis of AD-associated GWAS and OMIM genes and their first-degree interaction partners using forward genomics (GO-BP ITSS) may inform us about the relevance of protein–protein interactions, and corresponding domain–domain interactions. In figure 4B, we note that two AD genes (EPHA1 and CD2AP) interconnected through domain–domain interactions to two first-degree interactors (CBL and FYN) have potential biological roles in AD pathogenesis. EPHA1 (rs11767557; ephrin receptor A1) belongs to a subfamily of the protein tyrosine kinase family, and EPH receptors have been implicated in mediating developmental events, particularly in the nervous system. CD2AP (rs9349407; CD2-associated protein) encodes a scaffolding molecule that regulates the actin cytoskeleton. FYN is a member of the protein tyrosine kinase oncogene family and has been implicated in the control of cell growth. CBL is an oncogene that positively regulates receptor protein tyrosine kinase ubiquitination. CD2AP belongs to the cell projection organization biological process (figure 2) along with PVRL2 and PICALM (online supplementary table S3). In figure 4B, a candidate subnetwork of AD satisfies our three criteria of network modeling: (1) protein interactions between AD genes, (2) protein domains comprising the SNP, and (3) genetic association with AD. Consequently, the protein domains potentially associated with AD through known AD genes have been identified, and uncharacterized unique personal variants occurring in these regions are possibly more likely to be associated with AD than in other protein domains of these genes. Further, specific protein domains in a new gene are also imputed at higher probability of being associated with AD because they could affect the second-degree protein interaction between two AD genes.
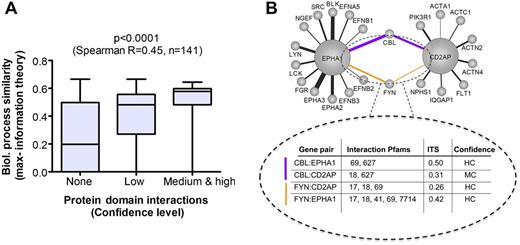
Triangulating at the nanoscale for prediction of unique personal variants: biological process similarity of Alzheimer's disease (AD) protein interactions correlates with the specific imputed protein domain-level mechanisms. (A) The confidence level of protein–domain interactions between genome-wide association studies (GWAS) and Online Mendelian Inheritance in Man (OMIM) AD genes, as well as first-degree interaction partners derived from the STRING database, were determined using the DMDM method (Materials and methods, DOMINE), and were significantly correlated with the similarity (information theory similarity (ITSS)) of higher-order Gene Ontology (GO) biological processes comprising these interactions using a non-parametric correlation (Materials and methods). (B) A subset of an AD GWAS protein–protein domain interaction network is shown with the specific domains responsible for the functional mechanisms underpinning AD imputed. Of note, only the protein interactions for which the imputed interacting protein domains are located in the GWAS or OMIM polymorphism are shown; in other words, protein interactions for which the protein domain could not be associated with an AD polymorphism located in the related genomic regions were filtered out. Large circles indicate AD genes (GWAS and OMIM), and small circles indicate first-degree interaction partners in STRING. Edge thickness is proportional to the ITSS between GO biological processes associated with these AD genes and their interactors (1≥ITSS≥0.193757; Materials and methods). The dotted circle highlights interconnected proteins and specific domains (Pfams) imputed as responsible for their interactions for two GWAS genes (EPHA1 and CD2AP) and two first-degree interactors (FYN, and CBL). Confidence levels: high confidence (HC); medium confidence (MC). ITSS scores (0–1) are listed for each gene pair, as well as confidence level ranges for domain interactions.
Limitations
We recognize several limitations in our work. First, we found that multiple subtypes of AD (eg, early- and late-onset, apraxia, familial types) may differ molecularly, although these disease traits share some clinical overlap. It is not clear whether there may be a bias in the autosomal form of AD, which may be very different from the complex inheritance forms, and therefore should not be mined as a cluster. Second, the intragenic SNPs may in fact correspond to genetic loci other than those currently annotated, and other, poorly understood, mechanisms may be at play. Third, we realize that the low yield for KEGG enrichment of AD genes may be due to low statistical power in terms of AD genes, KEGG pathway genes differentially expressed in AD rather than mutated, and/or because AD KEGG annotations are distributed across four distinct KEGG pathways, which may have compromised specificity in our analysis. Similarly, GO similarity and overlap analysis between OMIM and GWAS genes may have been limited by the quality and/or quantity of annotations within GO. Fourth, as noted in the Materials and methods section, because of the complex background of the GWAS SNPs, the enrichment studies of our 15 OMIM genes and 25 host genes of GWAS SNPs are below the conservative minimum number of genes for enrichment studies using theoretical statistics. Thus, we used empirical bootstrap statistics for our analysis, and further evaluated the robustness of the results by systematically removing one gene at a time from the enrichment study. Nonetheless, the observed results include high-level GO terms comprising thousands of less informative genes, which should be filtered out in future studies. Fifth, limitations in our protein-interaction modeling include the bias in the STRING database to annotate well-funded areas of research and the sensitivity to SPRING network quality cut-offs. Thus, we focused on a conservative cut-off, which yields fewer results. Also, cut-offs for the genes and relationships of interest are based on a scale-free model, which controls for hubs, but not for bottlenecks. There are many other ways to generate scale-free controls, and perhaps a model more balanced between hub and bottleneckness would be more insightful. Additionally, p values calculated on node degree and direct interaction may miss more subtle patterns. Related approaches for analyzing connections in more depth may be more biologically relevant. Finally, our Pfam analysis may have been limited by the fact that not all interactions or binding between proteins occur in Pfam domains. Future structural and biochemical work will largely inform future studies.
Conclusion
Implications for personal genomes
Here we have utilized forward phenomics methods and network models to automate predictions for biological mechanisms of AD inheritance at two different scales (protein interaction, GO terms); we thus imputed the ‘domain’ of intermediate systems (intermediate phenotypes) mediating the disease between molecular genetic levels and disease trait levels. We show that a significant amount of pathophysiologic connections are made between single and complex inheritance genes of AD at the GO level, although these biological constraints result in fewer relevant connections being made at the protein-interaction level. Importantly, we observe significant concordance between AD gene–domain interactions and protein interactions with AD GO-BP, which further constrain the potential disease-associated polymorphisms to a nanoscale subset of the protein region (figure 4). We propose that these AD-associated protein domains are thus predictive of complex or Mendelian disease inheritance as well as of new unique personal variants. Furthermore, these protein domains are associated in a functional genetic architecture which associates them with known disease-associated pathways. How we can further relate disparate connections between single and complex inheritance genes will be important in future studies for predicting personal variants and will likely require high-throughput predictions of protein structures of known or new polymorphisms occurring in these protein domains. Our proof-of-concept study holds promise for verifying and predicting unique personal variants in conjunction with known risk loci for AD, and may guide the interpretation of the forthcoming sets of deep-sequencing data for patients with AD, providing ‘prior hypotheses’ to reduce dimensionality in methods designed to analyze function in sequenced data.52 Significantly, by constraining at the mechanism level (protein–domain interaction, GO-BP), we are also able to extend inheritance information back to canonical knowledge of AD from KEGG (figure 5). Furthermore, our method is positioned to guide future studies identifying novel drug targets and drug repositioning methods in translating genetic findings into actionable targets of drug discovery, including protein domain networks confined within biological processes that extend known canonical pathways of AD. It is established that AD therapeutics will eventually require drugs targeted to the genomic aberrations underlying the disease.53 It has been shown that drug targets and disease genes coincide within PINs,54 and further methodology has been developed and independently validated to reposition drugs in this context, in addition to similarity measures of chemical components of drugs and molecular mechanisms.55–58 Such methods are well suited to integration of other high-throughput data utilized in our approach, including GO-BP and domain–domain interactions. Simultaneously, our method also effectively reduces dimensionality of biological mechanisms for this purpose (online supplementary table S9). Moreover, the greater weight of evidence of biological mechanisms compared with the original set of elementary SNPs for disease association queries would enhance the translation of these SNP–disease or personal variant–disease associations into existing drug networks using specific domain targets in the protein–protein interaction plane.
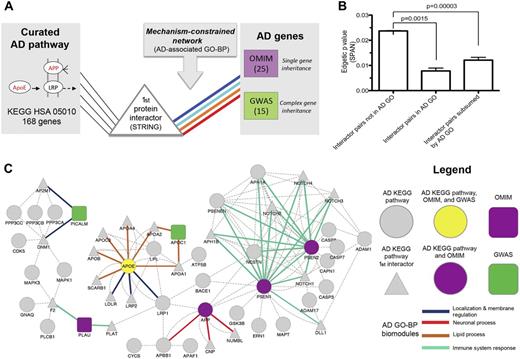
Extending known Alzheimer's disease (AD) pathways with genetic mechanisms (colored lines): indirect connections between single and complex inheritance AD genes and canonical KEGG AD pathway genes (hsa05010) constrained by AD-associated Gene Ontology (GO)-biological process (BP) mechanisms. AD polymorphisms of complex inheritance observed in genome-wide association studies (GWAS) are surprisingly genetically non-overlapping with the known pathway of AD and are established here as significantly interacting with the AD pathway (KEGG) via protein interactions that are themselves confined within established pathophysiological processes of AD (AD-associated GO-BP). (A) Outline of the process of discovering new AD mechanisms by extending known biological pathways of AD (genes of KEGG AD as circles) using first-degree protein-interaction partners (triangles, STRING network) selected according to their connectivity to the confirmed inheritable AD genes (squares, Mendelian gene in mauve (Online Mendelian Inheritance in Man (OMIM)) and complex (GWAS) in green), while simultaneously constraining at the biological process of these interactors to the same mechanism as that of the inheritable genetics (color of the interaction, shared GO-BP between interacting genes). In (B), we show that SPAN-prioritized protein interactions between the first interactor protein of the AD KEGG pathway and an AD gene (Mendelian or complex) are more likely to be observed between proteins that share an association with protein interaction to AD GO-BP mechanisms. Pairs of interacting proteins derived from the 40 AD genes and KEGG AD pathway (hsa05010) genes (STRING) that were not contained in AD GO terms (left bar), contained within the same shared AD GO term (middle bar), or subsumed by an AD GO class as defined in fig 2 (right bar) presented a more significant edgetic p value between their SPAN-modeled interactions than pairs not within AD GO (Kruskall–Wallis non-parametric analysis of variance p<0.0001; individual comparisons by Bonferroni-corrected Mann–Whitney test). Genes with interaction pairs sharing the same AD GO were found to be more significant than expected by chance (p<0.0001; 10 000 permutation resamplings of interactions between AD inheritance genes and AD KEGG first interactors). Conversely, a significant difference between interactor pairs containing the same shared AD GO and interactor pairs subsumed by an AD GO class was not observed. In (C), we show the protein-interaction network connecting KEGG AD pathway genes and their first interaction partners to AD GWAS and OMIM inheritance genes. Prioritized edges with p<0.05 (unadjusted p value of gene pair using empirical distribution from 10 000 permutation resamplings) within the protein–protein interaction network connecting AD GWAS and OMIM genes to genes and first interactors (STRING) from the KEGG AD pathway (hsa05010) are also constrained within four biomodules of GO-BPs found to be enriched in AD genes (see right bar of (B); online supplementary table S7 for interactor pairs mapped to AD GO-BP biomodules; online supplementary table S8 for interactor pairs mapped to shared AD GO-BPs). Node colors and shapes correspond to sources of genetic association (KEGG, OMIM, GWAS) and combinations thereof. Node shapes are hierarchical classes as follows: circle, canonical AD KEGG pathway gene; square, OMIM or GWAS AD inheritance gene; triangle, AD KEGG pathway first interactor. Node colors represent additional features following node shape: green, GWAS; purple, OMIM; gray, KEGG, first interactor to KEGG; yellow, KEGG, OMIM and GWAS. Each biomodule is color-coded according to each prioritized edge between KEGG AD first-degree interactor genes and AD inheritance genes. Of 103 interactions subsumed under AD-associated GO-BP, 750 met a SPAN network model of p<0.05 presented here, among 856 overall interactions identified between the AD KEGG pathways and the inheritable genes (single or complex). A larger network comprising first interactors from AD KEGG pathway genes connected to AD inheritance genes (428 interaction pairs) not constrained with respect to connections through AD KEGG pathway genes nor with an unadjusted p<0.05 cut-off was reduced to the network above using network modeling techniques. As expected, we find that GO-BPs common to both OMIM and GWAS connect to both forms of inheritance genes in the network. In this network, we found for the intersectional AD gene, APOE, among GWAS, OMIM and KEGG sources that three interactions with APOA1, APOA2, and APOA4 contain corresponding high-confidence domain–domain interactions mediated from a single protein domain of APOE, which also harbors 15 distinct AD-associated single-nucleotide polymorphisms (SNPs). APOE is mapped to the GO-BP protein–lipid complex remodeling (online supplementary table S4), whereby both high-density and low-density lipoprotein particle remodeling GO-BPs have plausible roles in the pathogenesis of AD (online supplementary table S5). Accordingly, APOA1, APOA2, and APOA4 all have roles in lipid metabolism, while APO1 and APO2 are the major components of high-density lipoproteins in the plasma. These protein and domain interactions may suggest how genetic aberrations among interaction partners can alter brain cholesterol metabolism and subsequently increase the risk of developing AD. Four other domain–domain interaction pairs not mapped to current AD-associated SNPs were also prioritized in this network (LRP1 with APP and APBB1; DNM1 with CDK5 and MAPK1).
Acknowledgments
We would like to acknowledge Ellen Rebman and James Chen for revisions.
Funding
This work was supported in part by K22 LM008308-04 (YAL-PI), 1K22CA143148 (MGK-PI), UL1RR029879, 5P30CA014599-35, 5UL1RR024999, by the Cancer Center of the University of Illinois in Chicago, and by the Institute for Genomics & Systems Biology at the University of Chicago. The funders had no role in study design, data collection, data analysis, decision to publish, or preparation of the manuscript.
Competing interests
None.
Contributors
Conceived and designed the experiments: MGK, YAL. Performed the experiments: KR, KW, ED, HL, JL, YL, MGK, YAL. Analyzed the data: KR, KW, ED, HL, JL, YL, MGK, YAL. Contributed reagents/materials/analysis tools: JL, YL, MGK, YAL. Wrote the paper: KR, KW, ED, HL, JL, YL, MGK, YAL. Conceived computational methods and interpreted the results: KR, KW, ED, HL, JL, MGK, YAL. Supervised the research: MGK, YAL.
Provenance and peer review
Not commissioned; externally peer reviewed.
References
Author notes
KR, KW, ED, HL and JL contributed equally to this work.
Work conducted in part at the University of Chicago.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}